Welcome to the Project MinE Databrowser
The Project MinE data browser is a web application designed to facilitate the exploration of genetic data related to amyotrophic lateral sclerosis (ALS). It provides an interactive platform to visualize and interpret genetic variants, and other relevant genomic information associated with ALS. The data presented in this browser is part of Project MinE. For more information on Project MinE visit the Project MinE website.
Browser Features


Datasets and Analyses
The current version of the browser includes a whole-exome sequencing dataset described in
Hop et al., 2026,
comprising 13,138 cases and 69,775 controls.
Three types of analyses are currently available:
- Single variant analysis: Exome-wide analysis and focused analysis of ALS-associated genes as defined by the ALS Gene Curation Expert Panel.
- Rare variant gene burden analysis: Burden analysis for ultra-rare variants, singletons, and ACAT tests at the gene level.
- Rare variant domain burden analysis: Burden analysis for ultra-rare variants, singletons, and ACAT tests at the protein domain level.
Overall, the Project MinE browser aims to advance our understanding of ALS genetics by providing a powerful and accessible tool for researchers and clinicians in the field.
For any questions regarding the Project MinE browser, feel free to reach out to projectmine@umcutrecht.nl.
Version: V1.0 | Last updated: March 31, 2026
Manhattan Plot
Genome-wide view of ALS associations for the different rare variant tests.
Association Table
Meta Table
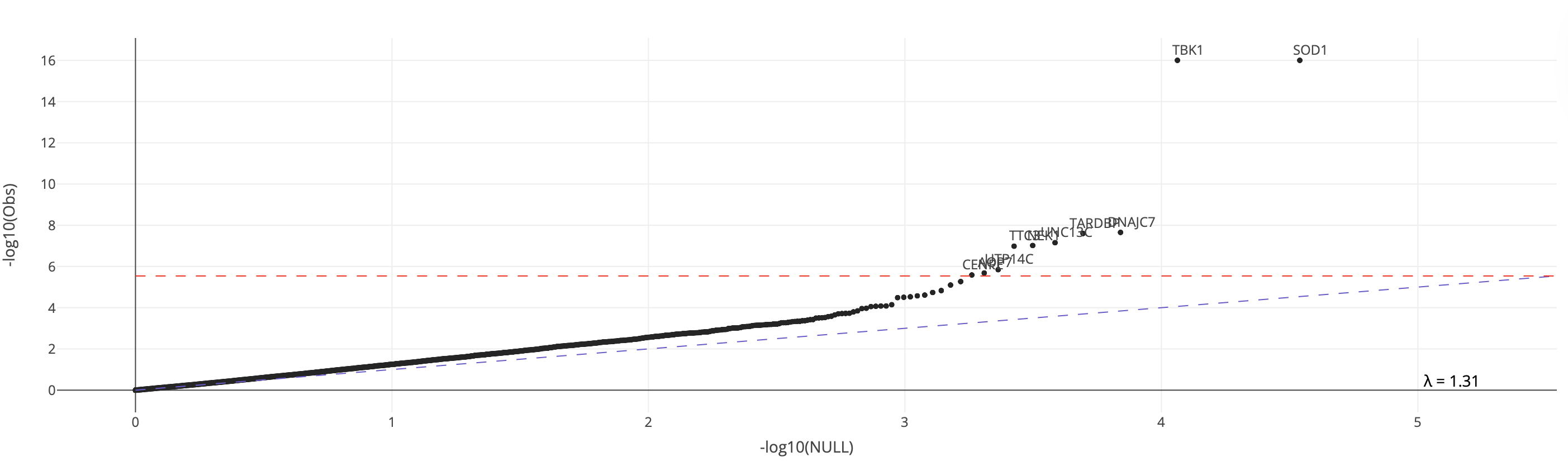
Quantile-Quantile plot
distribution of observed P-values relative to the null expectation
Association Table
Meta Table
Mutation Plot
Shows the distribution of rare variants (MAF < 0.05) in the selected gene, annotated with InterPro protein domains.
Variant table
Filter this table by clicking the variants in the mutation plot.
External Resources
About the Project MinE data browser
The Project MinE Databrowser is an open-access interactive platform that enables to explore gene-, domain-, and variant-level rare-variant associations in ALS, integrating statistical results with biological annotations to facilitate discovery and interpretation in an ALS-specific context. For more information on Project Mine, see the
Project MinE website.
For any questions regarding the Project MinE browser, feel free to reach out to projectmine@umcutrecht.nl.
The Whole-Exome Dataset
The whole-exome dataset is the largest ALS exome sequencing dataset to date, including 13,138 patients with ALS and 69,775 controls. All data is processed as described in Hop et al., 2026.
The Different Analyses
The different burden analyses consist of ultra-rare variants (≤5 carriers) or singletons. Additionally, variants were filtered based on predicted impact:
-
High Impact:
Nonsense mutations, splice acceptor/donor mutations, and frameshift mutations.
- Nonsense and frameshift mutations impact as predicted by snpEff. Splice variants as predicted by dbscSNV.
-
Moderate Impact:
Missense mutations, inframe deletions, and UTR truncations.
- Effect predicted by snpEff.
Tests across these filtering strategies were combined using the ACAT omnibus test.
For the burden analyses, both gene and domain coordinates were retrieved from Ensembl version 105.
Call Rate Filter
The super-cohort call-rate is a stringent filter that requires variants to have a call rate >0.9 in every super-cohort, ensuring reliable detection across datasets. Variants failing this threshold were excluded from the main exome-wide analysis. However, this stringent filter removes some known ALS-associated variants. Therefore, separate analysis of known ALS genes (GCEP) were performed without this super-cohort call-rate filter, allowing a more comprehensive investigation of these high-priority genes.
The super-cohorts are: WGS, WXS-UKB, and WXS-other. For more information see: Hop et al., 2026
Column Description
- Position: The variant's genomic position based on reference genome GRCh38 along with the other allele and effect allele.
- Impact: The variant's predicted impact based on snpEff and dbscSNV.
- Case MAF: The minor allele frequency of the variant in cases.
- Control MAF: The minor allele frequency of the variant in controls.
- Effect allele: The allele for which the effect size (e.g., OR or beta) is estimated.
- Other allele: The non-effect allele used as the reference in the association test.
- OR (95% CI): The odds ratio (OR) with its 95% confidence interval.
- P-value: Statistical significance of the association, calculated using the statistical test specified in the metadata table.
- Effect (95% CI): The effect estimate with its 95% confidence interval.
- Case MAC: The minor allele count in cases.
- Control MAC: The minor allele count in controls.
- Callrate cases: The proportion of cases with a non-missing genotype call.
- Callrate controls: The proportion of controls with a non-missing genotype call.
- Number of cases: The number of cases used in the association test.
- Number of controls: The number of controls used in the association test.
- rsID: The NIH's Reference SNP cluster ID.
- Transcript ID: The Ensembl base transcript ID.
- HGVSc: The HGVS coding DNA sequence name.
- HGVSp: The HGVS protein sequence name.
- dbscSNV Splicing variant: Specifies splice prediction as predicted by dbscSNV.
- GCEP classification: Gene-level evidence classification for ALS association, as defined by the ClinGen Gene Curation Expert Panel.
Burden-specific columns:
- Test Unit: Genomic region or functional unit (e.g., gene, exon, or region) over which variants are aggregated for burden testing.
- N variants: Number of variants included in the burden test for the given test unit.
- Start: The genomic position (GRCh38) where the test unit starts.
- End: The genomic position (GRCh38) where the test unit ends.
- Mean Case Burden: The average variant burden in cases.
- Mean Control Burden: The average variant burden in controls.
Version: V1.0 | Last updated: March 31, 2026